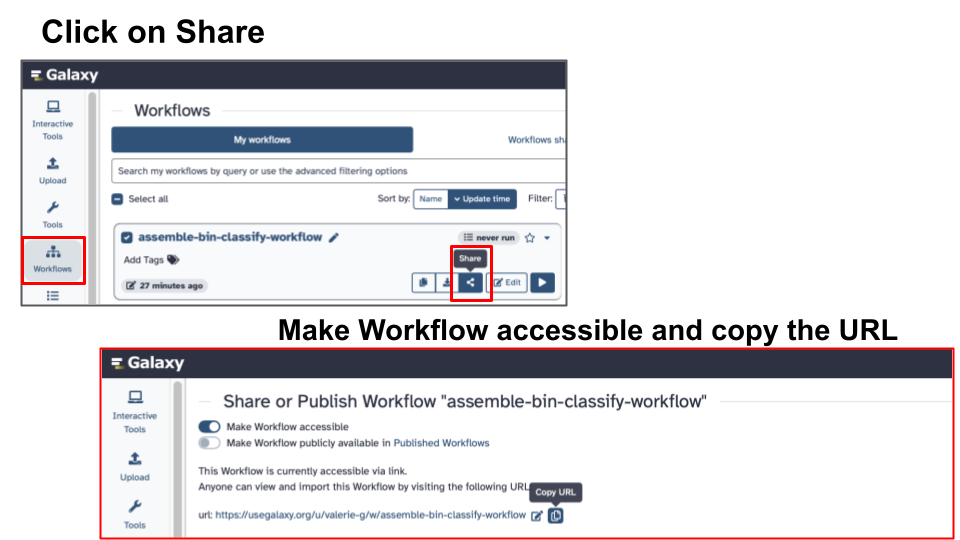
16.1 Galaxy Workflows: Build a Workflow
16.1.1 Purpose
A workflow is a reusable pipeline that chains tools and dataset actions so you can repeat the analysis consistently. Galaxy Workflows let you:
- Automate a series of analysis steps (tools + settings) so you don’t rebuild it each time.
- Ensure reproducibility by keeping the same order of steps and parameters when rerunning on new data.
- Scale and save time by running the same process across many samples consistently.
- Share and collaborate by exporting or sharing workflows so others can run the same analysis.
16.1.2 Learning Objectives
- Build a Galaxy Workflow from Scratch
- Run a Workflow
- Edit a Workflow
- Share a Workflow
16.1.3 Activity 1 - Create a Galaxy Workflow from scratch
Estimated time: 10 min
16.1.3.1 Instructions
Follow steps below to:
- Plan a Workflow - 3-tool workflow using Flye, MetaBAT2 and GTDB-Tk tools
- Create a Workflow - Start a new workflow
- Build a Workflow - Configure tool parameters, connect inputs/outputs
16.1.3.2 Step 1 - Plan a Workflow
- Use a high-quality, Zymo Gut Standard D6331 subset2:
| Tool | Goal | Specify Parameters | Required input type |
|---|---|---|---|
| Flye | De novo genome assembly | Mode: PacBio HiFi & metagenomic assembly | fastq |
| MetaBAT2 | Metagenomic binning | Default | fasta |
| GTDB-Tk | Classify genomes | Change versions to: Galaxy Version 2.5.2+galaxy1 | fasta (bins) |
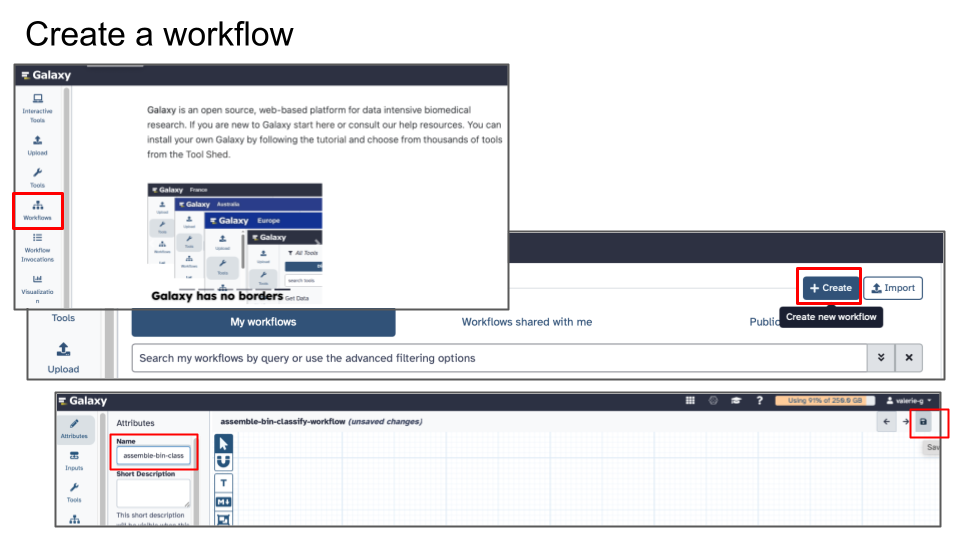
16.1.3.3 Step 2 - Create a Workflow
- In Galaxy’s menu on the Left, click Workflows
- Click Create new workflow
- Give it a name (e.g. pacbio-assemble-bin-classify)
- Click Save
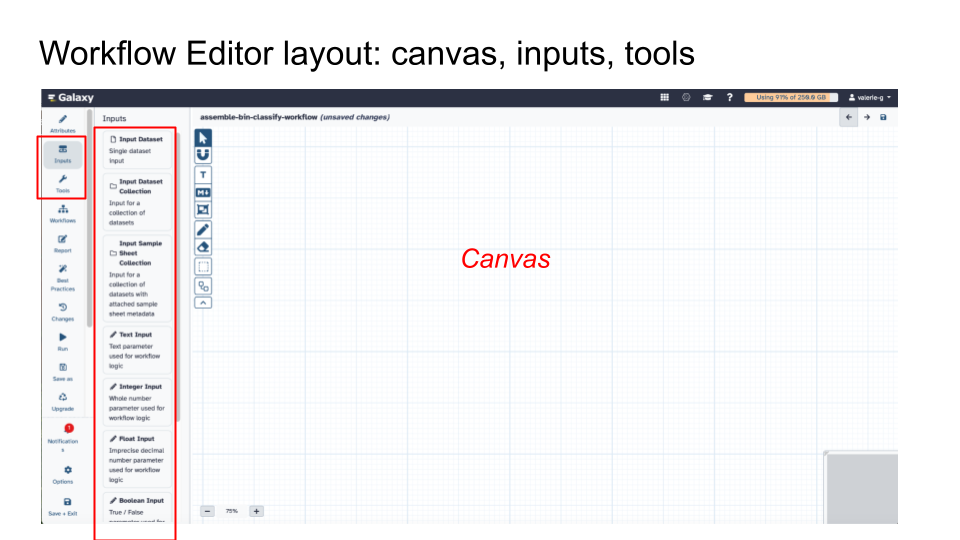
16.1.3.4 Step 3 - Build a Workflow
Note: While building your workflow you can save it at any point, but after saving, will need to click on Edit to go back and add.
In the Workflow Editor layout, on the canvas:
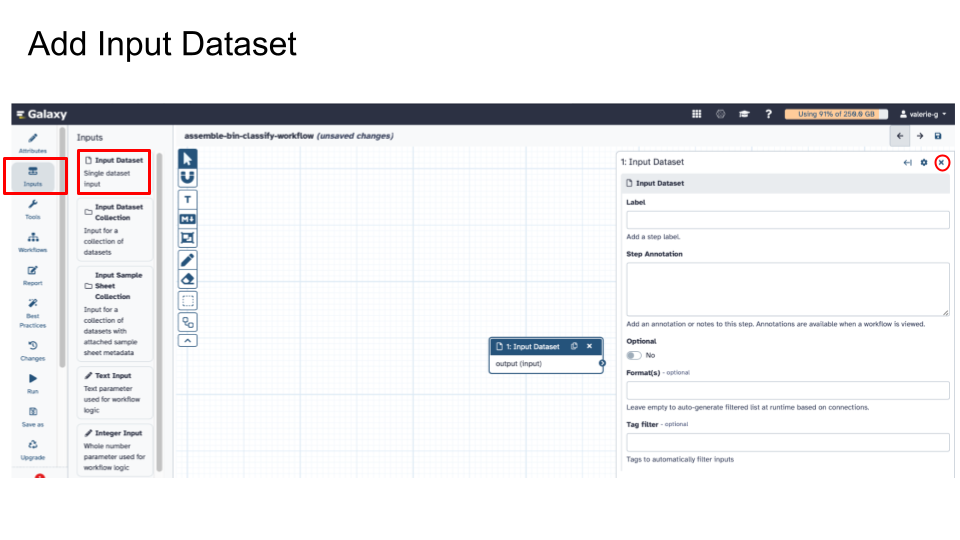
- Add Input Dataset (this is the workflow’s required input).
- For this activity, we are adding Single Dataset input as required input.
- For future workflows that you may run using more than one input file, you can specify Input Dataset Collection instead.
- Exit out of Input Dataset box to move on to another action.
- For this activity, we are adding Single Dataset input as required input.
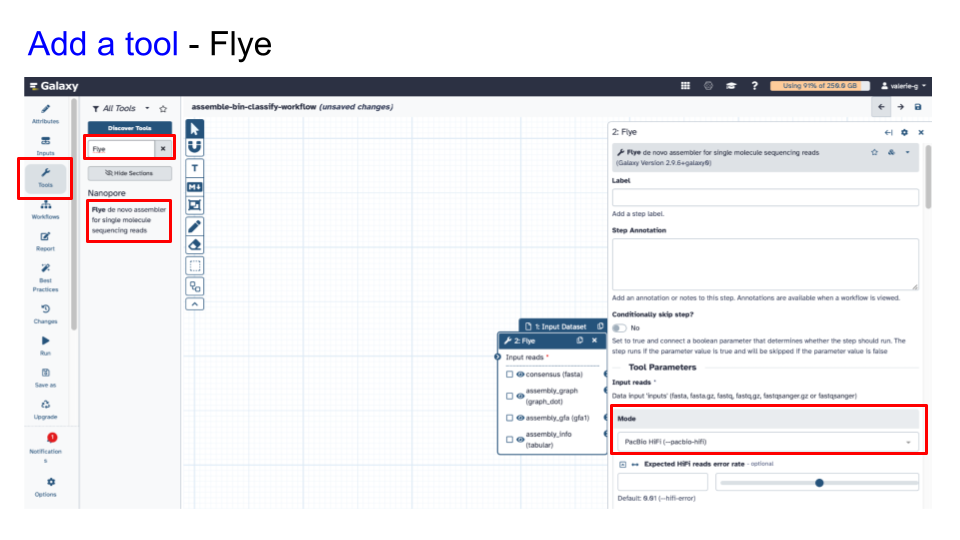
- Add tool #1 - Flye
- Specify parameters appropriate for your input
- Specify Mode: PacBio HiFi (–pacbio-hifi)
- Under Perform metagenomic assembly: select
Yes. - Under Generate a log file: select
Yes. - Leave rest as default
- Exit out of the Flye tool box
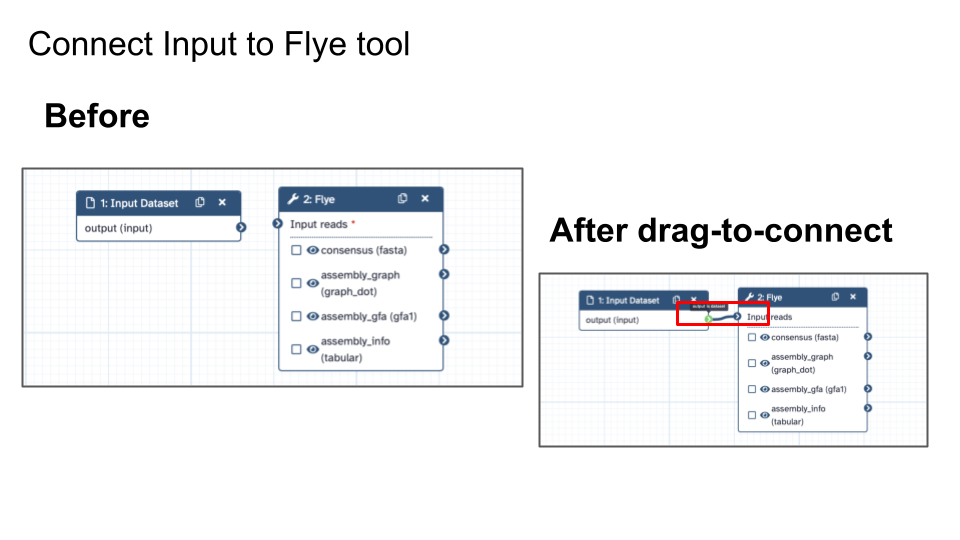
- Connect input/output
- On Workflow Canvas, reposition Input Dataset box and Tool box so it’s nice and comfortable for you to connect inputs and outputs
- Connect Input Dataset to Flye by dragging connector from Input Dataset to Flye.
- Specify parameters appropriate for your input
- Add tool #2 - MetaBAT2
- Specify parameters appropriate for your input
- Leave as default
- Exit out of the MetaBAT2 tool box
- Connect input/output
- On Workflow Canvas, reposition Input Dataset box and Tool box so it’s nice and comfortable for you to connect inputs and outputs
- Connect relevant output of Flye (consensus (fasta)), to MetaBAT2 by dragging connector from consensus (fasta) to MetaBAT2.
- Specify parameters appropriate for your input
- Add tool #3 - GTDB-Tk
- Specify parameters appropriate for your input
- Click on Versions button (next to the star) and Switch to 2.5.2+galaxy1 to be able to select an older GTDB-Tk database version
- Under GTDB-Tk database, select Full database - release 220 (2024-10-19)
- Leave rest as default
- Exit out of the GTDB-Tk tool box
- Connect input/output
- On Workflow Canvas, reposition Input Dataset box and Tool box so it’s nice and comfortable for you to connect inputs and outputs
- Connect relevant output of MetaBAT2(Bin sequences, fasta), to GTDB-Tk by dragging connector from bins to GTDB-Tk
- Click Save
- Specify parameters appropriate for your input
16.1.4 Activity 2 - Run Workflow
16.1.4.1 Instructions
- Import the dataset PacBio-sequenced Zymo gut standard D6331 subset:
- Go to Workflows, click to select your workflow and click Run
- Always double-check/select to ensure you are using the correct input file(s)
- USEFUL: check ‘Attempt to re-use jobs with identical parameters’. This option will allow you to re-run select steps of workflow if a step fails, without effectively re-running successful steps.
Note: It may take a few minutes to get the Workflow running e.g. time for loading and queuing steps.
16.1.5 Activity 3 - Modify Workflow
16.1.5.1 Instructions
- Add another tool (tool #4) to your workflow - add fastp!
- Think about where this tool fits in - why do we use it, and what would you do with fastp output?
- Think of a fastp parameter you might want to change to improve sequence quality.
- Think about input/output connections, and possible re-connections!
- When running the workflow, ensure you check ‘Re-use jobs with identical parameters’ to avoid re-running your other tools.